Dose Optimisation and Selection in clinical trials - Part 1: Trial Design

August 5, 2025
Author: Miguel Pereira
This is a 2-part article focusing on dose optimisation.
Part 1 will focus on elements of trial design as the basis to perform dose optimisation. In part 2, we will discuss statistical methodologies to analyse the data collected and carry out reliable dose selection.
Overview
In 2021, the FDA launched Project Optimus, a framework designed to provide guidance for improving dose optimisation in oncology drug development.
This initiative came on the back of the limitations of phase 1 dose-finding studies. These studies aim to identify the Maximum Tolerated Dose (MTD) and proceed by testing increasingly higher levels of a drug and defining the highest safe dose. This approach was valid for cytotoxic drugs where higher doses correlated with both higher toxicity and higher efficacy with a roughly linear relationship.
Why do we need dose optimisation for better dose selection?
Newer oncology drugs show non-linear dose-toxicity relationships. Moreover, there is not a good correlation between higher dose and higher toxicity and efficacy, with some drugs achieving an efficacy plateau before the MTD is reached (see image below).
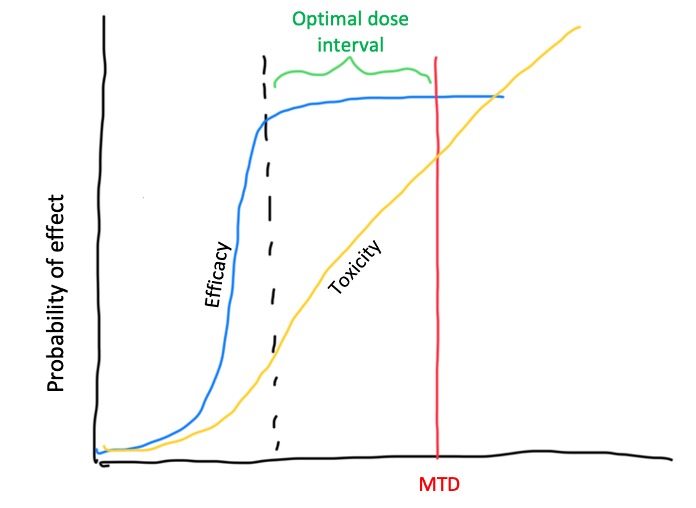
Additionally, methodologies used in phase 1 dose escalation have their own flaws. In particular, the 3+3 design, still the most commonly used methodology for dose-finding, find the right MTD only 33% of the time. This translates on a very small proportion of studies identifying the true MTD, among other pitfalls. Other methodologies, like the mTPI2 and BOIN designs, show better performance but still have caveats like the fact that decisions to escalate, stay and de-escalate are based on looking only at the current dose level while ignoring all the other data collected.
Side note: In my view, thinking about dose selection from an optimisation perspective, rather than the classical dose escalation perspective, is something that should be extended to all therapeutic areas. It’s not only oncology drugs that have novel mechanisms of action since innovation happens in all disease areas. Therefore, the generic principles described below should not be seen as exclusive to oncology although the FDA guideline applies only to oncology.
Project Optimus Guidance
There are two key points of project Optimus guidance that are important to mention:
- Randomization of patients - To minimise bias in the estimation of dose-toxicity and dose-response relationships
- Use of integrative analyses that include safety, tolerability, efficacy, pharmacokinetics (PK) and pharmacodynamics (PD) data.
The first point has implications on trial design, which are discussed below. The second has implications on the analysis which are discussed in part 2.
For all the details, the full guidance can be found here.
How to address Project Optimus?
Directions for Trial Design
Now that we have set the scene, how do we design a trial that addressed the requirements from Project Optimus?
Here are some directions about trial design with dose optimisation in mind:
- Phase 2 or phase 1/2 trials are the ideal settings to carry out dose optimisation (there are some directions below to add dose optimisation to phase 2/3 trials)
- Three dose cohorts should be considered (2 might be sufficient if clear differentiation exists). Testing 3 doses is advantageous because it allows the determination of a linear or non-linear dose-response/toxicity relationship
- Patient allocation to the 2-3 doses should be done by randomisation with, ideally, 20-30 patients per dose cohort. You can include data from phase 1 for the full analysis in a phase 1/2. For example, say you have seen 6 patients in each of the 3 dose levels that you want to test. You can ‘top-up’ each cohort with 14-24 patients/cohort randomised.
- Data from phase 1 should be considered in the analysis, including safety, efficacy and PK/PD.
- The diagram below shows possibilities for trial design and some of the analyses to be carried out:
How about doing dose optimisation in phase 3, registrational trials? This can be tricky because:
- Dose optimisation and selection adds uncertainty to the trial, as well as operational and statistical complexity
- If you are doing continuous enrolment, you need to consider the implications to enrolment while the dose selection is being carried out, namely:
- What happens to enrolment after all the data for dose selection is collected? Does enrolment continue or do you stop enrolment until you decide the dose? Assume at least 3 months from having all the data collected and making a decision on dose selection. This naturally impacts trial timelines.
- If you are planning a control arm (placebo or standard of care), do you start enrolling at the same time as you start the dose selection or only after dose selection is completed? This has implications on how you do sample size calculations and how you prepare the primary analysis of the phase 3 trial. This is particularly more complex when you have a time-to-event endpoint with an interim analysis.
- Additionally, note of caution, the Project Optimus guidance also states the following:

- This can add complexity to the statistical planning and analysis to set up an adaptive trial, whereby, one or more arms are dropped, and maintain type I error. This also means that the criteria for success might be more stringent for the primary endpoint, which is not desirable.
- Take-away: It is possible to design trials around all these possible options and contingencies but there is clearly more complexity that is worth considering before embarking on this path.
Wrap-up
While Project Optimus introduces additional complexity to trial design, the approach of exposing more patients to different doses early on and focussing identifying truly optimal rather than merely tolerable doses are a positive shift in clinical development.
In Part 2, we will discuss the analyses we can implement to tackle dose optimisation for regulatory submissions.